The Construction of a Novel Fusion Protein GSH-bFGF and the Characterization of Its Activities in vitro
Lin Chen1, Yi Zou1, Tianhong Zhou1, Hongjian Li1, Lin Cao2, *
1Department of Biology, School of Life Science and Technology, Jinan University, Guangzhou, China
2Department of Anesthesiology of Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, China
Email address

(Lin Cao)
*Corresponding author
Citation
Lin Chen, Yi Zou, Tianhong Zhou, Hongjian Li, Lin Cao. The Construction of a Novel Fusion Protein GSH-bFGF and the Characterization of Its Activities in vitro. International Journal of Chemical and Biomedical Science. Vol. 2, No. 2, 2016, pp. 9-16.
Abstract
The basic fibroblast growth factor (bFGF) and glutathione (GSH) have extensive biological functions. The bFGF is currently used to treat cardiovascular diseases, osteoarthritis, chronic kidney disease, Parkinson’s disease and mood disorders due to its mitogenic activity. Also, GSH deficiency has been linked to a number of human diseases such as liver cirrhosis, pancreatic inflammation, diabetes, neurodegenerative diseases, pulmonary diseases and aging. In this study, we construct a novel fusion protein, glutathione-basic fibroblast growth factor (GSH-bFGF) and characterized its bio-activities in vitro. The results showed that GSH-bFGF promoted the proliferation of cultured NIH/3T3 and primary human umbilical vein endothelial cells (HUVECs) using CCK-8 assays. The fusion protein also displayed well preserved antioxidant activity using Total Antioxidant Capacity Assay and significantly inhibited the senescence of HUVECs. We therefore, proposed that comparing with GSH or bFGF alone, the synthetic novel fusion protein GSH-bFGF was likely to have improved potentials in treating aging related diseases, such as atherosclerosis and neurodegenerative diseases.
Keywords
GSH-bFGF, Proliferation, Antioxidant Ability, Cell Senescence, Atherosclerosis
1. Introduction
Fibroblast growth factor was first isolated in 1974, from bovine pituitary gland and was shown to have mitogenic effect on murine fibroblast Balb/c 3T3 cell line [1]. Basic fibroblast growth factor (bFGF) is a member of a large family of structurally related multipotential glycoproteins that promote the proliferation, migration, differentiation and the survival of various cell types [2].
The bFGF was purified by heparin affinity chromatography in 1980’s as the first potent endothelial cells mitogen [3, 4]. The molecular weight of bFGF is 18 kDa, while larger forms of bFGF (22, 22.5 and 24 kDa) resulted from alternate CUG-translation start sites have also been identified [5]. The bFGF contains four cysteine residues with no intramolecular disulfide bonds, a large number of basic residues (pI 9.6) and two sites (Ser 64 and Thr 112) can be phosphorylated by protein kinases A and C, respectively [6]. bFGF acts through binding and activation of specific fibroblast growth factor receptors, which are tyrosine kinase receptors containing three immunoglobulin-like domains, a single-pass transmembrane domain and a cytoplasmic tyrosine kinase domain [7, 8]. FGFR isoforms (FGFR1b, FGFR1c, FGFR2c, FGFR3c and FGFR4) are synthesized by expressions of splice variants of a given FGFR gene or by expressions of different FGFR genes. Alternative splicing specifies the sequence of the carboxy-terminal half of the Ig domain III, resulting in either III b or III c isoforms [9].
Two FGF molecules connected by a heparan sulfate proteoglycan bind to extracellular IgII and IgIII domains of FGFR and lead to receptor homodimerization [10]. The stimulation of FGFRs activates several signaling pathways, amongst which the best understood is Ras–mitogen-activated protein kinase (Ras/MAPK) pathway, phosphoinositide 3-kinase–Akt (PI3K/AKT) pathway and Phospholipase C-γ (PLCγ) pathway [11]. Receptor activation leads to tyrosine phosphorylation of FGFR substrate 2 (FRS2), which serves as an essential core upon which a signaling complex consisting of the tyrosine phosphate Shp2, the adaptor Grb2, and the docking protein Gab1 is formed [12]. Formation of this signaling complex subsequently activates Ras/MAPK and PI3K/AKT pathways [13]. It is noteworthy that activation of PI3K /AKT pathway leads to phosphorylation of MDM2, which then translocate to the nucleus and degrades p53 subsequently. p53 is a transcription factor of p21, which inhibits cell growth and maintain cell cycle arrest via inhibiting a number of cyclins and cyclin-dependent kinases [14].
Given the mitogenic activity of bFGF, bFGF stimulates cell growth, cell migration, cell differentiation, cell survival and angiogenesis and therefore, has been widely used in regenerative medicine. Recent researches also revealed that bFGF functioned downstream of the aging suppressor klotho and endogenous bFGF expression was downregulated with aging [14]. Therapeutic targeting of bFGF signaling components has been shown to reverse some of the cellular processes of aging [15]. In fact, various FGFs are currently tested for therapeutic applications of a number of age-related disorders such as cardiovascular diseases, diabetes, osteoarthritis, chronic kidney disease, Parkinson’s disease and mood disorders [9].
The glutathione (GSH), γ-L-glutamyl-L-cysteinylglycine, is a ubiquitous low-molecular-mass non-protein biothiol in plant and animal cells [16]. GSH is a key element of the antioxidant defense system and plays vital roles in detoxification and in protecting cells against reactive oxygen species. In recent years, protein glutathionylation has also been recognized as an effective post-translational modification to regulate target proteins activities [17]. The cellular functions of GSH include: 1) detoxifying electrophiles; scavenging free radicals; 2) maintaining the essential thiol status of proteins; 3) providing a reservoir for cysteine; 4) modulating critical cellular processes such as DNA synthesis, microtubular-related processes, and immune function [18]. Therefore, GSH deficiency has been shown to relate to a number of human diseases such cancer, liver cirrhosis, cardiovascular diseases, neurodegenerative diseases and aging [19].
Given the mitogenic activity of bFGF and the antioxidant activity of GSH and the crosstalk of both pathways in regulating cell proliferation, differentiation and aging, we constructed a novel fusion protein GSH-bFGF in this study. Purified GSH-bFGF was subsequently tested for its potential antioxidant activity and mitogenic activity in vitro. The results showed that the novel fusion protein well maintained antioxidant capacity and mitogenic activity. GSH-bFGF significantly promoted the proliferation of cultured cells and inhibited the senescence of the cultured primary HUVEC cells. Furthermore, GSH-bFGF fusion protein likely functioned via PI3K /AKT pathway since downregulated expression of p21 was observed in GSH-bFGF induced cells. Further characterizations of the biological activity of this novel fusion protein in vivo will hopefully provide more understanding on the potential application of GSH-bFGF in treating aging related conditions.
2. Materials and Methods
2.1. GSH-bFGF and bFGF Expression Constructs
Total mRNA was extracted from spleen using Trizol reagent according to the manufacturer’s protocol and was subsequently transcribed into cDNA. The complete cDNA of bFGF was amplified with a primer pair (5’-GATCATATGGCTGCTGGTA-3’ and 5’- ACTGAGATCTTCAGCTCTT-3’) and cloned into pET21b vector. The pET21b-GSH-bFGF was subsequently constructed with a primer pair containing 3 tandem glutathione codons upstream bFGF ORF (5’-GATCATATGGAATGCGGTGAGTGTGGCGAATGCGGTATG-3’ and 5’-ACTGAGATCTTCAGCTCTT-3’) using pET21b-bFGF as a template. PCR was performed in a Peltier Thermal Cycler 200 programmed with an initial denaturation at 94°C for 5 minutes followed by 35 cycles of denaturation at 94°C for 30 seconds, annealing at 50°C for 1 minute and extension at 72°C for 40 seconds. The PCR products were verified by electrophoresis on 1% agarose gel and visualized by ethidium bromide staining under ultra violet (UV) light. The PCR products were then purified from gels and cloned into pET21b vector to generate the expression constructs of bFGF and GSH-bFGF, respectively. The sequences of the constructs were verified by DNA sequencing (Ruibo Biotech, Beijing).
2.2. Expression of bFGF and the Novel Fusion Protein GSH-bFGF
pET21b-GSH-bFGF and pET21b-bFGF were used to transfect E. coli BL21, respectively. The transfected cells were incubated at 37°C overnight in LB supplemented with 100 μg/ml ampicillin under shaking. The overnight cell cultures were used to inoculate fresh LB containing 100 μg/ml ampicillin, and then cultivated at 25°C until the OD600 of the media reached 0.8. The protein expressions were then induced with 1mM isopropyl-d-thiogalactopyranoside (IPTG; Fermentas) at 25°C for 7 hours under shaking.
The above cell cultures were harvested using centrifugation at 10,000 rpm for 10 minutes at 4°C. The cell pellets were then resolubilized in 1% w/v lysis buffer (NaH2PO4·2H20 50 mmol/L, pH 8.0; NaCl 300 mmol/L, imidazole 10 mmol/L) and incubated on ice for 30 minutes. The lysate was further solubilized using ultrasound sonication at brief intervals at 4°C. The cell lysate were then spined at 12,000 rpm at 4°C for 10 minutes and the supernatant was collected for subsequent protein purification.
2.3. Purification of Fusion Protein GSH-bFGF and bFGF
The target proteins containing six his tags were first purified by Ni-NTA Fast Kit (GE healthcare). ~80 ml cell lysate containing the overexpressed target proteins were loaded and the columns were washed with 5 column volumes washing buffer (NaH2PO4·2H20 50 mmol/L, pH 8.0; NaCl 300 mmol/L; Imidazole 20 mmol/L) to remove nonspecific proteins. The fusion proteins that remained on the column were then washed off with 10 ml elution buffer (NaH2PO4·2H20 5 mmol/L, pH 8.0; NaCl 300 mmol/L; Imidazole 250 mmol/L). Ten fractions of 1 ml were collected and 20 µl aliquots of each fraction were preserved for further analysis by SDS-PAGE.
The protein samples were further purified by cation exchange chromatography using SP high performance prepacked column (GE healthcare). The above fractions containing the peak elution of target proteins were pooled and were then dialyzed against PBS at 4°C overnight. The volumes of the protein solutions were finally reduced to 1 ml using ultrafiltration. The samples were loaded on SP high performance prepacked column at 1 ml/min and were washed at 2 ml/min with phosphate buffer. The fusion proteins were then eluted at 1 ml/min with gradient salt buffer containing NaCl up to 1 mol/L (prepared with 1mol/L NaCl and PB according to the manufacturer’s instruction). Protein elusion was monitored using AKTA purifier 10 (GE healthcare) and target proteins were observed when NaCl was > 400 mmol/L. Agian, 10 fractions of 1ml containing the peak elution of target proteins were collected and 20 µl aliquots of each were preserved for further analysis by SDS-PAGE. The protein concentration was determined by Bicinchoninic acid method.
2.4. Immunoblotting Analysis
Protein samples were mixed with 2× loading buffer (0.125 M pH 6.8 Tris-HCl, 4% w/v SDS, 0.004% w/v bromophenol blue, 20% glycerol, and 10% β-mercaptoethanol), and heated at 100°C for 10 minutes. Protein samples were separated on 12% SDS-PAGE gel and subsequently western transfered to nitrocellulose membranes, performed as described in Current Protocols in Cell Biology. The membranes were then immunoblotted with appropriate primary monoclonal antibody and horseradish peroxidase conjugated secondary antibodies. The target proteins were finally detected by Enhanced Chemiluminescence (Amersham).
2.5. Cell Proliferation Assay
The NIH/3T3 cells were cultured in DMEM medium (Hyclone) supplied with 10% FBS (Hyclone), 100 units/ml penicillin and 100 μg/ml streptomycin (Gibco). The primary HUVECs were cultured in Endothelial Cell Medium (Sciencell) containing 5% FBS, 1% ECGS, 100 units/ml penicillin and 100 μg/ml streptomycin. The cultured primary HUVECs after 10 passages were recognized as senescent HUVECs in our study. For cell proliferation assay, cells were harvested at 90% confluence and diluted to 2.5×104/ml. 2500 cells (~100 μl per well) were plated on 96-well plate and cultured for additional 24 hours. The medium were then replaced with DMEM or ECM containing 0.5% FBS and GSH-bFGF or bFGF at various concentrations for additional 48 hours. The cell proliferations were monitored using Cell Counting Kit-8 (CCK-8, Dojindo, Shanghai) following the manufacturer’s instruction. The optical density values at 450 nm were measured for each sample and normalized against the values of blank control. For long-term cell culture, ~ 2500 primary HUVECs were used to inoculate 100 μl Endothelial Cell Medium (Sciencell) containing 5% FBS, 1% ECGS, 100 units/ml penicillin, 100 μg/ml streptomycin with or without 10 ng/ml GSH-bFGF/bFGF. Cells were harvested every four days and cell numbers were counted. Only 2500 cells were kept to inoculate 100 μl culture medium again. The population doubling times were calculated as the logarithm two of the times of cell multiplication and at each time point, the population doubling times were the sum of the current and all the previous population doubling times.
2.6. Antioxidant Activity Analysis
The potential antioxidant activity of GSH-bFGF was determined using Total Antioxidant Capacity Assay (T-AOC Assay Kit, Beyotime biotechnology). 5% Oxidant ABTS+ was added to Trolox that diluted to 0.15, 0.3, 0.6, 0.9, 1.2, 1.5 mmol/L, respectively, and the reactions were allowed for 6 minutes at room temperature. The optical density values at 745 nm were monitored and were used to construct a standard curve, as described by the manufacturer. The antioxidant activities of samples were calculated according to the standard curve.
2.7. Cell Aging Analysis
The senescence of cultured HUVECs was monitored using Cell Senescence Assay Kit (Beyotime technology, Shanghai), following the manufacturer’s instructions. Briefly, 5×104 cells were plated on 3.5 cm dish and cultured to 30-35% confluence before further analysis. The cells were washed three times with PBS and were then fixed in 1 ml fixing solution (PBS mixed with 2% formaldehyde and 0.2% glutaraldehyde) for 5 minutes at room temperature. The cells were subsequently washed with 3 ml PBS three times and then subjected to SA-β-gal staining at 37°C overnight. The cells were then imaged using an inverted microscope (Nikon TS100).
2.8. Data Analysis
Values are shown as mean ± SEM in the text and figures. Differences between the groups were analyzed using one-way analysis of variance, followed by Bonferroni test. Probability values less than 0.05 were considered significant.
3. Results
3.1. Construction and Expression of Fusion Protein GSH-bFGF
Total mRNA was isolated from spleen using Trizol reagent according to the manufacturer’s protocol. The complete cDNA of bFGF was amplified by RT-PCR and cloned into pET21b vector. The pET21b-GSH-bFGF was subsequently constructed with pET21b-bFGF using a 5’ primer containing 3 glutathione codons upstream bFGF ORF and therefore, the three glutathione were at the N-terminal of bFGF in the novel fusion protein GSH-bFGF. The sequences of both constructs were verified using sequencing (Ruibo Biotech, Beijing).
The pET21b-GSH-bFGF and pET21b-bFGF vectors were used to transfect E. coli BL21 (DE3) competent cells, respectively. High expressions of GSH-bFGF fusion protein and bFGF were observed with 1 mmol/L IPTG induction at 25°C (Figure 1).
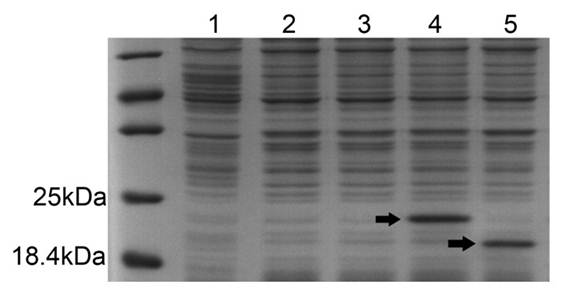
Figure 1. The expression of GSH-bFGF and bFGF in E. coli BL21 (DE3) competent cells.
pET21b-GSH-bFGF or pET21b-bFGF were used to transfect E. coli BL21 (DE3), respectively. ~10 μg total soluble proteins in cell lysate were separated on 12% SDS-PAGE, followed by coomassie brilliant blue staining. High expressions of GSH-bFGF (~20.4 kDa) and bFGF (~19.3 kDa), indicated with arrows, were observed with 1mmol/L IPTG induction.
Lane 1: total proteins of E. coli BL21 transfected with control plasmids. Lane 2: total proteins of E. coli BL21 (uninduced) transfected with pET21b-GSH-bFGF. Lane3: total proteins of E. coli BL21 (uninduced) transfected with pET21b-bFGF. Lane 4: total proteins of E. coli BL21 (induced with 1 mmol/L IPTG) transfected with pET21b-GSH-bFGF. Lane 5: total proteins of E. coli BL21 (induced with 1 mmol/L IPTG) transfected with pET21b-bFGF. The protein marker was loaded on the lane left to lane 1.
3.2. Purification and Verification of GSH-bFGF Fusion Protein
Cloned into pET21b, both GSH-bFGF and bFGF were expressed with six his-tags at C terminus and were then subjected to nickel affinity chromatography using Ni-NTA Fast Kit. ~15 ml cell lysate of BL21 overexpressing GSH-bFGF or bFGF were loaded on a nickel-chelating resin and the columns were washed with the washing buffer containing 20 mM of imidazole to eliminate the nonspecific proteins. The target proteins were then eluted with 10 ml (10 column volumes) elution buffer containing 250 mM of imidazole. 10 fractions of 1 ml were collected and 20 μl aliquots of each fraction were separated on 12% SDS-PAGE. The gel was then stained in coomassie brilliant blue for 1-2 hours on a platform rocker and a few contaminants as well as target protein were observed (Figure 2A).
Both GSH-bFGF (pI 9.07) and bFGF (pI 9.6) were positively charged in pH 7.45 phosphate buffer and supposed to bind cation exchange column. The above fractions that were rich of target proteins were pooled and were further purified by cation exchange chromatography. The target proteins were subsequently eluted with gradient salt buffer containing NaCl up to 1 mol/L. The target proteins were first appeared in the elutes when the concentration of NaCl in the elusion buffer was increased up to 400 mmol/L. Again, 10 fractions of 1 ml were collected and 20 μl aliquots of each fraction were again separated on 12% SDS-PAGE. The gels were then stained in coomassie blue and the contaminants were minimized (Figure 2B, 2C).
The above purified GSH-bFGF fusion protein and bFGF were again separated on 12% SDS-PAGE and verified by immunoblotting using anti-bFGF monoclonal antibody. Specific band of GSH-bFGF (~ 20.4 kDa) and bFGF (~19.3 kDa) were observed (Figure 2D). The protein concentrations were determined by bicinchoninic acid protein quantification assay and ~0.2 mg GSH-bFGF and ~0.4 mg bFGF were finally recovered.
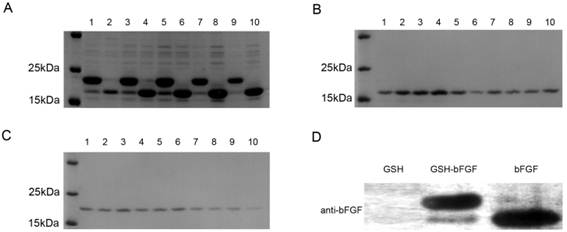
Figure 2. Purification of GSH-bFGF and bFGF using nickel-chelating resin, followed by cation exchange chromatography.
Protein samples from each fraction collected were separated on 12% SDS-PAGE and visualized using coomassie blue staining, monitored using immunoblot with indicated specific antibody.
A: Target proteins were purified using Ni-NTA from the cell lysate of BL21 transfected with pET21b-GSH-bFGF (lane 1, 3, 5, 7, 9) or pET21b-bFGF (lane 2, 4, 6, 8, 10), respectively. The peak elution of GSH-bFGF was observed in fraction 1, 3, 5, 7 while the peak elution of bFGF was observed in fraction 4, 6, 8, 10, respectively.
B: The above fractions containing large amount of bFGF were pooled and were subjected to cation exchange chromatography. Contaminants were minimized in the protein elusions.
C: The above fractions containing large amount of GSH-bFGF were pooled and were subjected to cation exchange chromatography. Contaminants were minimized in the protein elusions.
D: The purified protein samples of bFGF and GSH-bFGF were separated on 12% SDS-PAGE and verified using monoclonal anti-bFGF. A ~20.4 kDa band for GSH-bFGF and a ~19.3 kDa band for bFGF were shown as expected, GSH was used as a negative control.
3.3. GSH-bFGF Fusion Protein Displayed Antioxidant Activity in vitro
Glutathione has long been known for its antioxidant activity that acts alone or in concert with enzymes in cells to reduce free radicals, which play essential roles in lots of human diseases such as cancer, cardiovascular disease, renal disease and neurodegenerative diseases, as well as in senescence. The potential antioxidant activity of GSH-bFGF fusion protein was determined in vitro using Total Antioxidant Capacity Assay Kit with a rapid ABTS method (T-AOC Assay Kit, Beyotime biotechnology) in this research. Trolox was used to construct a standard curve, as described by the manufacturer. The antioxidant capacity of purified GSH-bFGF was measured and compared with that of Trolox and was recorded as Trolox Equivalent Antioxidant Capacity. Commercially manufactured GSH (Wako) was used as a positive control. Our results showed that the antioxidant activity of GSH-bFGF was (5.36±0.3) mM and the commercial GSH was (4.72±0.07) mM.
3.4. The Potential Effect of GSH-bFGF Fusion Protein on Cell Proliferation
bFGF has long been shown to stimulate the proliferation of different cells both in vitro and in vivo. In this study, the potential effect of the novel fusion protein GSH-bFGF on cell proliferation was determined in cultured NIH/3T3 fibroblasts as well as in cultured senescent HUVECs (Figure 3). The cells were maintained in culture medium containing GSH-bFGF or bFGF at various concentrations for 48 hours and the cell proliferation was monitored using CCK-8 assay. bFGF was used as the positive control. Comparing with blank control, significantly improved cell proliferations were observed for both GSH-bFGF and bFGF treatment at only 10 ng/ml (Figure 3). In addition, the mitogenic effects of both GSH-bFGF and bFGF displayed dose-dependent manner ranging from 10 ng/ml to 100 ng/ml. Interestingly, GSH-bFGF displayed stronger mitogenic potentials than bFGF at all tested dosages, although no significant differences were observed. The ED50 for GSH-bFGF was 10 ng/ml.
Similar results were observed in cultured senescent primary HUVECs, which express endogenous bFGF and respond further to this growth factor added exogenously [20]. In addition, as shown in figure 3B, GSH-bFGF further increased cell proliferation at high dosage (> 100 ng/ml), while bFGF displayed decreased mitogenic activity at high concentration (> 100 ng/ml). The data indicated an elevated mitogenic activity for GSH-bFGF, possibly due to the combined effects of its antioxidant activity and the mitogenic activity.
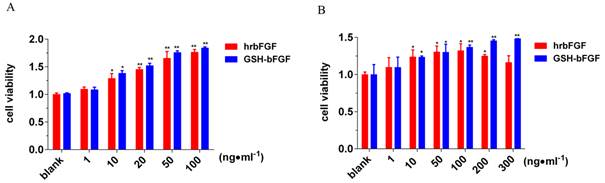
Figure 3. The influence of GSH-bFGF on cell proliferation.
Purified GSH-bFGF or bFGF was used to treat cultured NIH/3T3 cells (A) and cultured senescent HUVECs (B). The potential impact of GSH-bFGF and bFGF on cell proliferation was monitored using CCK-8 assay and normalized against the blank control. Both GSH-bFGF and bFGF significantly increased cell proliferation in a dose-dependent manner, ranging from 1 ng/ml to 100 ng/ml. In addition, the mitogenic activity of bFGF decreased at high concentration (> 100 ng/ml), while the mitogenic activity of GSH-bFGF was further increased at 200 ng/ml and 300 ng/ml. * P<0.05, **P<0.01. Culture medium only was used in the blank control.
3.5. The Long-Term Effect of GSH-bFGF on Cell Proliferation
The mitogenic activity of GSH-bFGF was further determined in long-term cell culture using primary HUVECs. ~2500 cells were used to inoculate 100 μl medium per well and the cells were maintained in culture medium containing 10 ng/ml of GSH-bFGF, 10 ng/ml of bFGF (positive control) or medium only (blank control). The cells were harvested and counted every four days. Only 2500 cells were kept and used to reinoculate 100 μl medium per well. The cell population doubling times were then calculated and the population doubling times of previous passages were added. Our results showed that the proliferation of control primary HUVECs were arrested after 40 days (10 passages) while the proliferations were still observed at 52 days (13 passages) on cells maintained in GSH-bFGF or bFGF (Figure 4). GSH-bFGF displayed elevated mitogenic potential at the later stage of long-term cultured HUVECs (aging HUVECs, >10 passages), in consistent with our previous results.
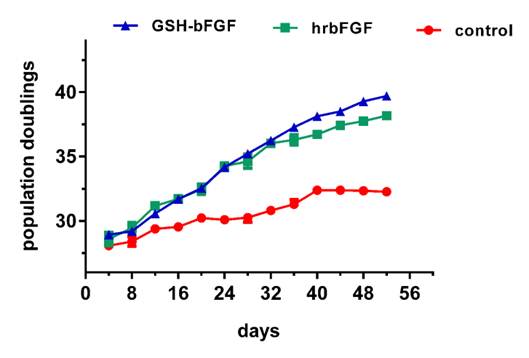
Figure 4. GSH-bFGF displayed long-term mitogenic activity in cultured cells.
The long-term effects of GSH-bFGF and bFGF on cell proliferation were determined using cultured primary HUVECs. The growth curves of long-term cultured primary HUVECs were constructed using the population doubling times calculated every four days. Increased cell proliferation of HUVECs was observed with 10 ng/ml of GSH-bFGF and 10 ng/ml of bFGF treatment. Also, both GSH-bFGF and bFGF inhibited cell senescence since the control cells stopped dividing after 10 passages while cell proliferation was still observed with GSH-bFGF/bFGF treatment after 13 passages. Mock treatment using culture medium was used as blank control. *P<0.05, **P<0.01.
3.6. The Novel Fusion Protein GSH-bFGF Inhibited Cell Senescence in Cultured Endothelial Cells
Given the antioxidant activity of GSH and the mitogenic activity of bFGF, the potential impact of the fusion protein GSH-bFGF on cell senescence was determined using senescent primary HUVECs. At the end of the above long-term cell culture (52 days, 13 passages), cell senescence of primary HUVECs was monitored using Cell Senescence Assay Kit. The color reaction of X-gal, which representing the elevated senescence-associated β-galactosidase (SA-β-gal) expression in aging cells, was obviously less in GSH-bFGF or in bFGF treated cells, comparing with that in control cells (Figure 5). The results indicated the inhibited senescence in GSH-bFGF and bFGF treated cells.
The inhibition of senescence induced by GSH-bFGF was further elaborated by showing that p21 was significantly dowregulated in cells subjected to GSH-bFGF or bFGF treatment (Figure 5D). In general, inhibited cell proliferation in senescent cells was mediated by CDKi (cyclin-dependent kinase inhibitors). Our results suggested that GSH-bFGF and bFGF might accelerate cell proliferation and inhibit cell senescence via p21 signaling pathway.
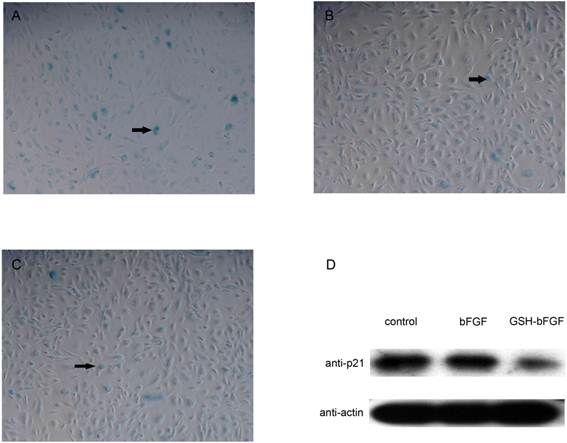
Figure 5. GSH-bFGF inhibited the expression of p21 and the cell senescence of HUVEC cells.
At the end of the long-term cell culture (after 13 passages, 52 days cell culture) using HUVECs, the senescence of cultured HUVECs was monitored using SA-β-gal staining (blue stains, as indicated with arrows). Comparing with the control cells (A), the blue stain was significantly decreased in cells treated with 10 ng/ml bFGF (B) as well as in cells treated with 10 ng/ml GSH-bFGF (C). The expression levels of p21 in the above cells were further determined using western blot with specific monoclonal anti-p21 antibody (D). The expression of p21 was inhibited in cells treated with 10 ng/ml bFGF and was further inhibited in cells treated with GSH-bFGF. Mock treatment using culture medium only was used in the blank control.
4. Discussion
Cell senescence (replicative senescence) was described as the limited number of cell divisions of normal cells, accompanied with the altered cell morphology and gene expressions. Cell senescence can also be induced (stress-induced premature senescence) by various stress factors, such as radiation, oxidative stress and inflammations. Both types of cell senescence share similar mechanisms and display common hallmarks, predominantly via activating p53 and its downstream effector p21 and p16, which are inhibitors of cyclin-dependent kinases and mediate cell cycle arrest [21].
Cell senescence underlies various health conditions such as neurodegenerative disorders, coronary heart disease, post-trauma recovery and cancer. Basic fibroblast growth factor (bFGF) has long been known for its mitogenic activity, which promotes the cell proliferation and migration and improves cell survival of many different cell types both in vitro and in vivo [22, 23, 24]. Endogenous bFGF expression levels increases during development and gradually decreases with aging [25]. The tripeptide, GSH which protect redox-related enzymes and (or) proteins from reactive oxidant species (ROS) and reactive nitro species (RNS), which play pivotal roles in cell senescence [26]. Decreased GSH levels are also one of the common features of aging cells [27]. In this study, we constructed a novel fusion protein GSH-bFGF and characterized its biological functions in vitro. The fusion protein GSH-bFGF maintained its antioxidative activity in vitro, comparing with GSH only. Also, compared with bFGF, GSH-bFGF displayed improved mitogenic activity in cultured NIH/3T3 cells and senescent HUVECs. Furthermore, better tolerance has been observed in senescent HUVECs by showing that GSH-bFGF further stimulated cell proliferation at high dosages (>100 ng/ml), while decreased cell proliferation was observed with bFGF at the equivalent concentrations. The compromised mitogenic activity of bFGF supplementation at high dosage could be the consequence of downregulated expression of FGF receptor at high cell density, due to the initially accelerated cell proliferation [28]. The mitogenic activity of GSH-bFGF was likely to be the combined effect of the activation of growth factor pathway and the antioxidant activity, which eventually improved cell survival and reduced apoptosis.
In our long-term cell culture using HUVECs, both GSH-bFGF and bFGF significantly stimulated cell proliferation. The control cells stopped dividing after 10 passages while the cell proliferation were still observed with the cells maintained in 10 ng/ml GSH-bFGF or in 10 ng/ml bFGF. Comparing with bFGF only, GSH-bFGF displayed improved mitogenic activity particularly on senescent HUVECs (after 10 passages) in our long-term cell culture, possibly due to the antioxidant activity of GSH, which eventually reduced the free radicals increasingly accumulated in aging cells and protected cells from aging. This hypothesis was further confirmed in cell senescence assay using SA-β-gal staining and GSH-bFGF inhibited the senescence of cultured primary HUVECs significantly. In consistent, significantly decreased expression of p21 was observed in the GSH-bFGF treated HUVECs. However, no change of the expression of p53 in GSH-bFGF treated senescent HUVECs was observed in our study (data not shown). Therefore, GSH-bFGF might inhibit cell aging via signaling pathways other than MDM2/p53, or this might be cell type specific since several groups showed that the levels of p53 protein do not rise in some senescence cells [29].
5. Conclusion
In this study, we constructed a novel fusion protein GSH-bFGF. The fusion protein well preserved the antioxidant activity of GSH and the mitogenic activity of bFGF in vitro. Also, the fusion protein GSH-bFGF was well tolerated in our long-term cell culture using primary HUVECs and significantly inhibited the cell senescence. On the other hand, the clinical application of GSH as anti-oxidative and anti-aging drugs has been restricted because of its low bioavailability. The novel fusion protein GSH-bFGF may help the intake of GSH through FGFRs. Further investigations using different types of cells such as chondrocytes and neuron cells and in vivo assays will be helpful to understand the protective effects and the potential application of this fusion protein in treating aging related disorders.
Acknowledgement
This project was founded by National Natural Science Foundation of China (31301190) and Science and Technology Planning Project of Guangdong China (2013B051000023).
References
- Gospodarowicz D. Localisation of a fibroblast growth factor and its effect alone and with hydrocortisone on 3T3 cell growth [J]. Nature, 1974, 249: 123-127.
- Acevedo V D, Ittmann M, Spencer D M. Paths of FGFR-driven tumorigenesis [J]. Cell Cycle, 2009, 8(4): 580-588.
- Maciag T, Mehlman T, Friesel R, et al. Heparin binds endothelial cell growth factor, the principal endothelial cell mitogen in bovine brain [J]. Science, 1984, 225(4665): 932-935.
- Shing Y, Folkman J, Sullivan R, et al. Heparin affinity: purification of a tumor-derived capillary endothelial cell growth factor [J]. Science, 1984, 223 (4642): 1296-1299.
- Prats H, Kaghad M, Prats A C, et al. High molecular mass forms of basic fibroblast growth factor are initiated by alternative CUG codons [J]. Pro Natl Acad Sci, 1989, 86 (6): 1836-1840.
- Bikfalvi A, Klein S, Pintucci G, et al. Biological Roles of Fibroblast Growth Factor-2 1 [J]. Endocr Rev, 1997, 18 (1): 26-45.
- Mohammadi M, Olsen S K, Ibrahimi O A. Structural basis for fibroblast growth factor receptor activation [J]. Cytokine Growth F R, 2005, 16 (2): 107-137.
- Yun Y R, Won J E, Jeon E, et al. Fibroblast growth factors: biology, function, and application for tissue regeneration [J]. J Tissue Engineering, 2010, 1(1): 218142.
- Johnson D E, Lu J, Chen H, et al. The human fibroblast growth factor receptor genes: a common structural arrangement underlies the mechanisms for generating receptor forms that differ in their third immunoglobulin domain [J].Mol Cell Biol, 1991, 11 (9): 4627-4634.
- Schlessinger J, Plotnikov A N, Ibrahimi O A, et al. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization [J].Mol Cell, 2000, 6(3): 743-750.
- Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy [J]. Nat Rev Drug Discov, 2009, 8(3): 235-253.
- Sternberg P W, Alberola-Ila J. Conspiracy theory: RAS and RAF do not act alone [J]. Cell, 1998, 95 (4): 447-450.
- Dailey L, Ambrosetti D, Mansukhani A, et al. Mechanisms underlying differential responses to FGF signaling [J]. Cytokine Growth F R, 2005, 16 (2): 233-247.
- Coutu D L, Galipeau J. Roles of FGF signaling in stem cell self-renewal, senescence and aging [J]. Aging (Albany NY), 2011, 3 (10): 920-33.
- Jin K, Sun Y, Xie L, et al. Neurogenesis and aging: FGF-2 and HB‐EGF restore neurogenesis in hippocampus and subventricular zone of aged mice [J]. Aging cell, 2003, 2 (3): 175-183.
- Penninckx M J, Elskens M T. Metabolism and functions of glutathione in micro-organisms [J]. Adv Microb Physiol, 1993, 34: 239-301.
- Ghezzi P. Protein glutathionylation in health and disease [J]. BBBA Gen Subjects, 2013, 1830 (5): 3165-3172.
- Meister A, Anderson M E. Glutathione [J]. Annu Rev Biochem, 1983, 52 (1): 711-760.
- Wu G, Fang Y Z, Yang S, et al. Glutathione metabolism and its implications for health [J]. J Nutr, 2004, 134 (3): 489-492.
- Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature, 1993, 801-809.
- Fridlyanskaya I, Alekseenko L, Nikolsky N. Senescence as a general cellular response to stress: A mini-review [J]. Exp Gerontol, 2015, 72: 124-128.
- Suzuki T, Akasaka Y, Namiki A, et al. Basic fibroblast growth factor inhibits ventricular remodeling in Dahl salt-sensitive hypertensive rats [J]. J Hypertens, 2008, 26 (12): 2436-2444.
- Tomanek R J, Zheng W, Yue X. Growth factor activation in myocardial vascularization: therapeutic implications [J]. Mol Cell Biochem, 2004, 264 (1-2): 3-11.
- Kanazawa T, Komazawa D, Indo K, et al. Single injection of basic fibroblast growth factor to treat severe vocal fold lesions and vocal fold paralysis [J]. The Laryngoscope, 2015, 125 (10): E338-E344.
- Liu Y, Yi X C, Guo G, et al. Basic fibroblast growth factor increases the transplantation‑mediated therapeutic effect of bone mesenchymal stem cells following traumatic brain injury [J]. Mol Med Rep, 2014, 9 (1): 333-339..
- Colavitti R, Finkel T. Reactive oxygen species as mediators of cellular senescence [J]. IUBMB life, 2005, 57 (4-5): 277-281.
- Homma T, Fujii J. Application of Glutathione as Anti-Oxidative and Anti-Aging Drugs [J]. Curr Drug Metab, 2015, 16(7): 560-571.
- Nakamizo S, Egawa G, Natsuaki Y, et al. Topical treatment with basic fibroblast growth factor promotes wound healing and barrier recovery induced by skin abrasion [J]. Skin Pharmacol Physiol, 2013, 26 (1): 22-29.
- Webley K, Bond J A, Jones C J, et al. Posttranslational modifications of p53 in replicative senescence overlapping but distinct from those induced by DNA damage [J]. Mol Cell Biol, 2000, 20 (8): 2803-2808.